
Wat is het Chitayat-syndroom?
Het Chitayat-syndroom is een erfelijke aangeboren aandoening waarbij pasgeboren kinderen problemen hebben met ademhalen in combinatie met meerdere bijzondere uiterlijke kenmerken.
Hoe wordt het Chitayat-syndroom ook wel genoemd?
Chitayat is een arts die dit syndroom beschreven heeft in 1993.Het wordt ook afgekort met de letters CHYTS.
ERF-gerelateerd syndroom
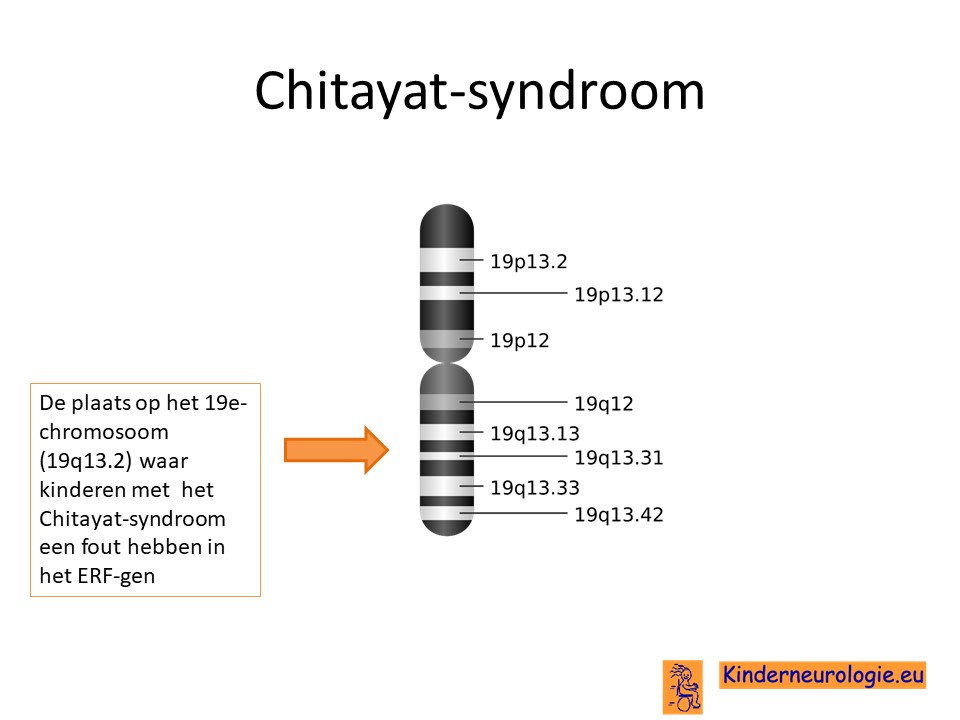
Een andere naam die ook gebruikt wordt is het ERF-gerelateerd syndroom. ERF-gen is de naam van de plaats in het erfelijk materiaal (het DNA) waar kinderen met dit syndroom een fout hebben zitten die verantwoordelijk is voor het ontstaan van de symptomen die horen bij dit syndroom. Door deze naam te gebruiken is het voor iedereen duidelijk dat er sprake is van deze fout. Een syndroom is een combinatie van kenmerken en problemen die een zelfde oorzaak hebben.
Craniosynostose type 4
Een fout op dezelfde plek in het DNA als bij het Chitayat syndroom kan ook alleen zorgen voor een veranderd vorm van de schedel zonder de andere symptomen die horen bij het Chityat syndroom. Deze veranderde vorm van de schedel wordt craniosynostose genoemd. Er bestaan verschillende fouten in het DNA die allemaal een craniosynostose kunnen veroorzaken. Deze vorm wordt type 4 genoemd.
Chitayat-Hall syndroom
Er bestaat ook een syndroom dat het Chitayat-Hall syndroom wordt genoemd, dit is een ander syndroom dan het Chitayat syndroom. tegenwoordig wordt het Chitayat-Hall syndroom ook wel het Schaaf-Yang syndroom genoemd.
Hoe vaak komt het Chitayat-syndroom voor bij kinderen?
Chitayat-syndroom is een zeldzaam voorkomende aandoening. Het is niet goed bekend hoe vaak deze aandoening voorkomt bij kinderen. De aandoening is sinds 1993 bekend als aandoening. Geschat wordt dat deze aandoening bij minder dan één op de 100.000 kinderen voorkomt.
Waarschijnlijk is bij een deel van de kinderen die het Chitayat-syndroom heeft, de juiste diagnose nog niet gesteld, omdat de aandoening nog niet herkend is. Dankzij nieuwe genetische technieken is het gemakkelijker geworden deze diagnose te stellen, waardoor er meer duidelijkheid zal komen hoe vaak deze aandoening nu daadwerkelijk voorkomt.

Bij wie komt het Chitayat-syndroom voor?
Het Chitayat-syndroom is al voor de geboorte aanwezig. Meestal wordt kort na de geboorte duidelijk dat er ademhalingsproblemen zijn in combinatie met meerdere bijzondere uiterlijke kenmerken.
Zowel jongens als meisjes kunnen dit syndroom krijgen.
Wat is de oorzaak van het ontstaan van het Chitayat-syndroom?
Fout in het erfelijk materiaal
Het Chitayat-syndroom wordt veroorzaakt door een fout in het erfelijk materiaal. Deze fout ligt op chromosoom 19. De plaats van de fout in het erfelijk materiaal wordt het ERF-gen genoemd.
Autosomaal dominant
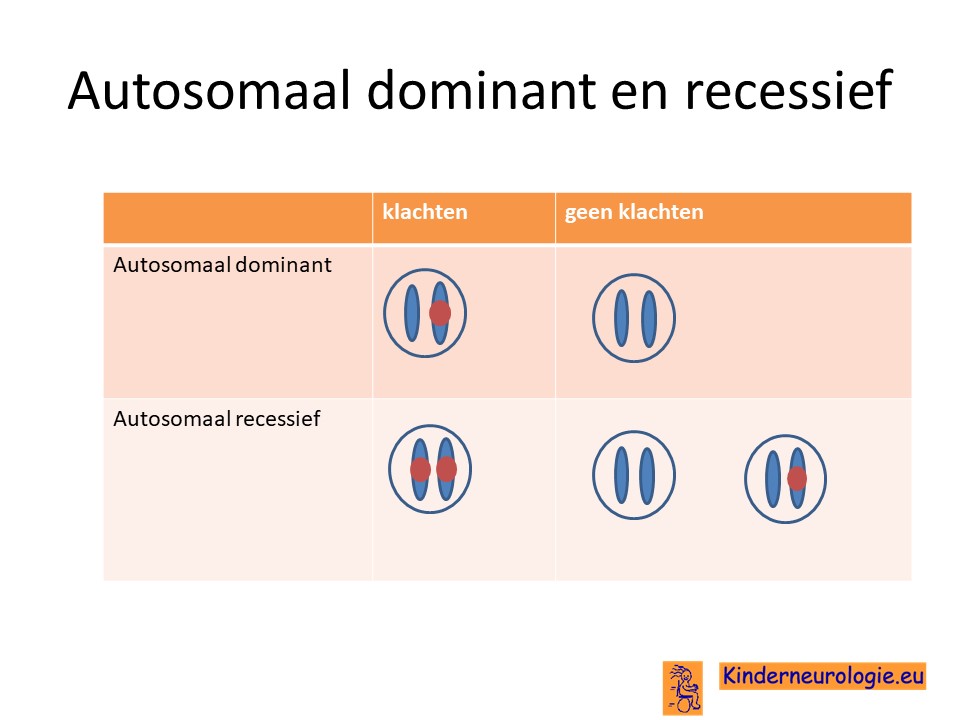
Het Chitayat-syndroom erft vaak op een zogenaamd autosomaal dominante manier over. Dit houdt in dat een fout op één van de twee chromosomen 19 die een kind heeft, al voldoende is om klachten te krijgen. Dit in tegenstelling tot autosomaal recessieve overerving waarbij een kind pas klachten krijgt wanneer beide chromosomen 19 een fout bevatten.
Bij het kind zelf ontstaan
Bij een groot deel van de kinderen met het Chitayat-syndroom de fout in het DNA bij het kind zelf ontstaan bij de bevruchting van de eicel door de zaadcel. De vader of de moeder hebben zelf niet het Chitayat-syndroom. Dit wordt de novo genoemd, wat nieuw ontstaan bij het kind betekent.

Geërfd van een ouder
Een ander deel van de kinderen heeft de fout in het DNA geerfd van een ouder die zelf ook het Chitayat-syndroom heeft. Soms was dit al bekend, soms wordt de diagnose pas gesteld wanneer de diagnose bij het kind gesteld wordt.

Type fout in het DNA
Het type fout in het DNA bepaalt ook hoeveel en welke klachten een kind zal gaan krijgen. Bepaalde fouten zorgen er voor dat er helemaal geen werkzaam eiwit meer wordt gemaakt, deze fouten hebben de grootste gevolgen voor een kind. Bij ander type fouten wordt er een afwijkend maar nog wel werkzaam eiwit gemaakt of is er minder goed werkzaam eiwit dan gebruikelijk. Deze kinderen zullen minder klachten hebben dan kinderen die helemaal geen werkend eiwit meer hebben.

Afwijkend eiwit
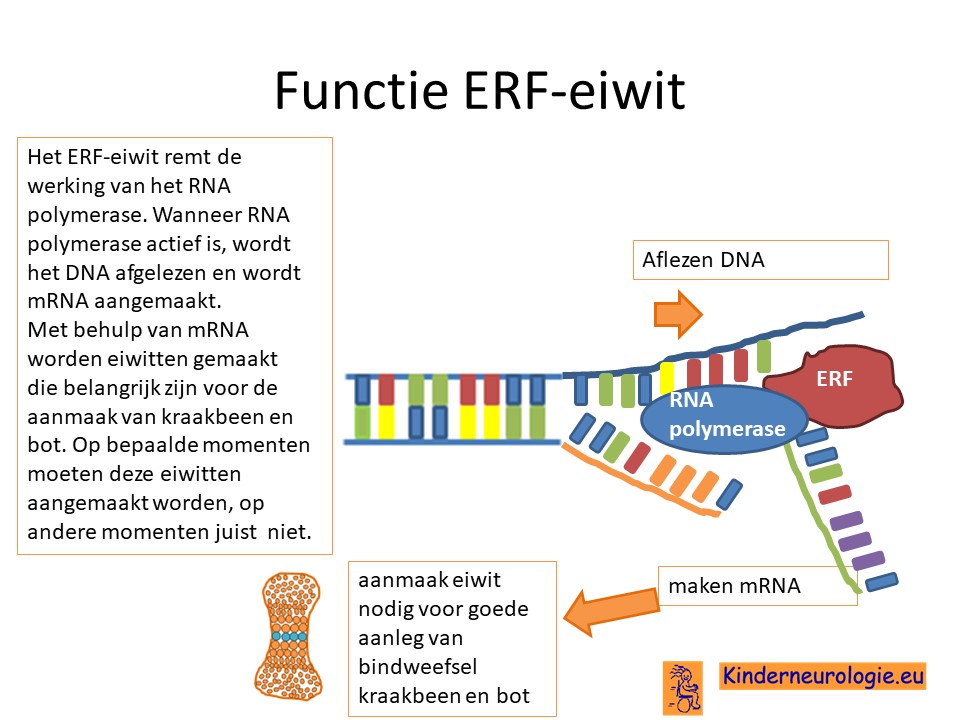
Als gevolg van de fout in het Chitayat-gen wordt een bepaald eiwit,het ETS repressor factor (ERF-eiwit) niet goed aangemaakt. Dit eiwit speelt een belangrijke rol bij het aflezen van het DNA tijdens de ontwikkeling van een kind in de baarmoeder. Het eiwit speelt vooral een belangrijke rol bij de aanmaak van kraakbeen en van bot. Door een afwijking in dit eiwit wordt bindweefsel, het kraakbeen en bot anders aangelegd dan gebruikelijk waardoor een deel van de symptomen die horen bij dit syndroom ontstaan.
Luchtpijp
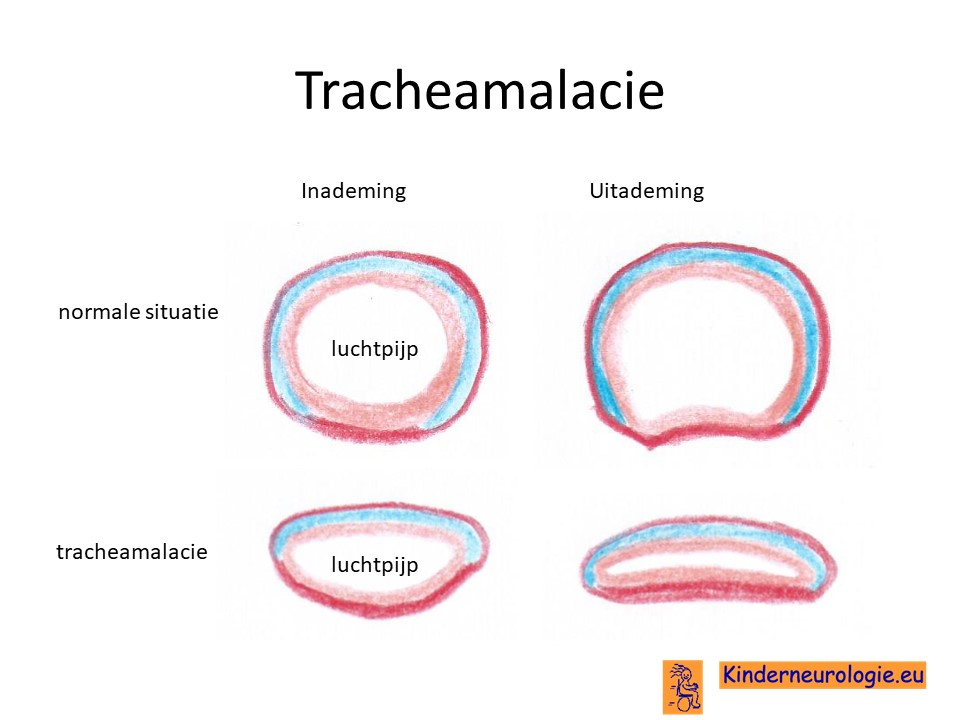
In de wand van de luchtpijp zit kraakbeen, waardoor de luchtpijp stevigheid krijgt en zijn ronde vorm krijgt. Bij kinderen met het Chitayat syndroom is dit kraakbeen van minder goed kwaliteit, waardoor de luchtpijp veel smaller en slapper is dan gebruikelijk. De luchtpijp beweegt mee tijdens de ademhaling en kan tijdens de uitademing helemaal dichtvallen. Dit geeft problemen met ademhalen.
Longen
Dit eiwit speelt ook een belangrijke rol bij de aanleg van de longen. Bij kinderen met het Chitayat syndroom blijken de longblaasjes minder mooi rond van vorm te zijn en ook groter te zijn dan gebruikelijk. Ook kan er meer bindweefsel tussen de longblaasjes aanwezig zijn. Op deze manier kunnen de longblaasjes minder goed zuurstof uit de lucht opnemen en minder goed koolzuur aan de lucht afgeven.

Craniosynostose
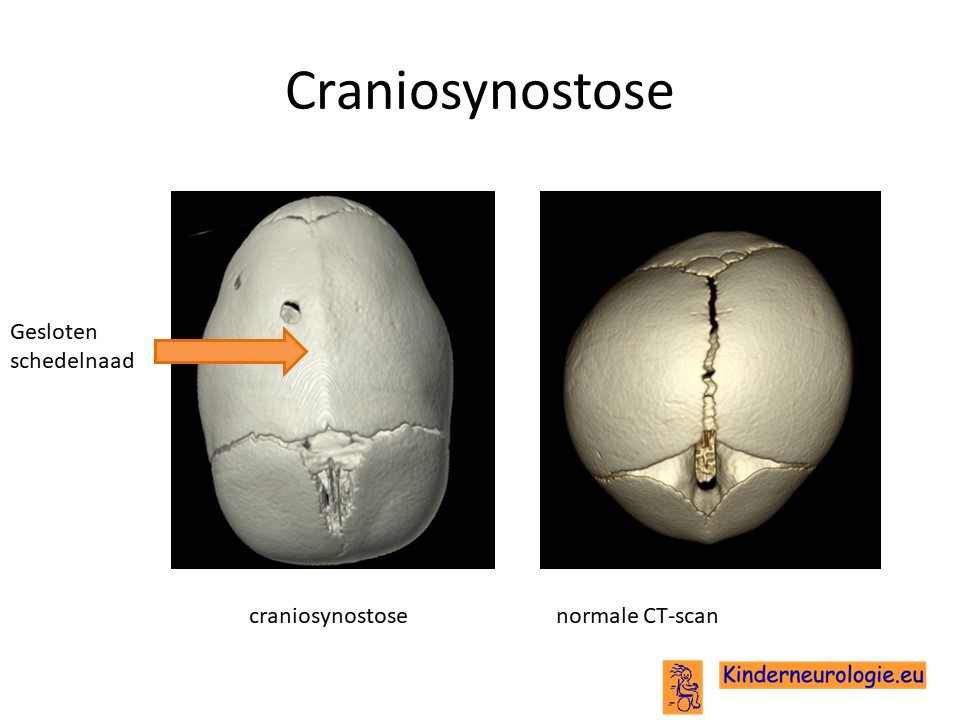
Bij een pasgeborene bestaat de schedel uit meerdere lossen stukken. Tussen deze botdelen liggen de schedelnaden. Deze schedelnaden gaan geleidelijk aan naar elkaar toegroeien. Doordat de schedelnaden nog niet aan elkaar vast zitten, kan het hoofd van een pasgeborene en dreumes gemakkelijk groter worden. Wanneer een of meerdere van de schedelnaden te vroeg aan elkaar vastgroeien, kan de schedel niet op de gebruikelijke manier groeien en ontstaat een afwijkende vorm van de schedel. Dit wordt een craniosynostose genoemd. Door de fout in het ERF-gen sluiten de schedelnaden te vroeg, waardoor kinderen met het Chitayat syndroom vaak een afwijkende vorm van de schedel hebben.

Wat zijn de symptomen van het Chitayat-syndroom?
Variatie
Er bestaat variatie tussen de hoeveelheid en de ernst van de onderstaande symptomen die verschillende kinderen met het Chitayat-syndroom hebben. Het valt van te voren niet te voorspellen welke symptomen een kind zal krijgen. Dat betekent dat onderstaande kenmerken kunnen voorkomen, maar niet hoeven voor te komen.

Jouw kind is uniek
Bedenk dat onderstaande symptomen kunnen voorkomen bij jouw kind, maar ook niet allemaal zullen voorkomen. Jouw kind is uniek en veel meer dan een kind met deze aandoening. Het lezen van mogelijke symptomen die kunnen voorkomen, kan ouders het gevoel geven dat er alleen maar aandacht is voor de beperkingen van het kind. Dat is zeer zeker niet de bedoeling. Jouw kind is bijvoorbeeld lief, grappig, gevoelig, gezellig,sociaal, vindingrijk, nieuwsgierig, ondeugend, enthousiast,een zonnestraaltje, creatief en/of innemend en dat vind je niet terug in onderstaande symptomen die kunnen horen bij dit syndroom. Dat kan ook niet, want die eigenschappen maken jouw kind nu eenmaal uniek. Blijf daar vooral naar kijken en zie deze symptomen meer als achtergrondinformatie die je kunnen helpen om te begrijpen wat er met je kind aan de hand zou kunnen zijn wanneer jouw kind zich anders ontwikkelt of ergens last van heeft. Deze informatie kan jullie als ouders en hulpverleners een handvat geven wat hiervoor een mogelijke verklaring kan zijn.

Zwangerschap en bevalling
Een deel van de moeders heeft tijdens de zwangerschap een vergrote hoeveelheid vruchtwater. Hierdoor is de buik van de moeder groter dan te verwachten is op grond van de zwangerschapsduur. Een te grote hoeveelheid vruchtwater wordt polyhydramnion genoemd.
Lage spierspanning
Een deel van de babys' met het Chitayat-syndroom heeft een lage spierspanning. Deze kinderen voelen na de geboorte slapper aan dan gebruikelijk en moeten meer ondersteund worden wanneer ze opgetild worden. Vooral de spieren van de nek hebben een lage spierspanning waardoor kinderen moeite hebben om hun hoofd overeind te houden. Kinderen met deze aandoening zijn soepel in hun gewrichten, deze kunnen gemakkelijker overstrekt worden.

Problemen met ademhalen
Een groot deel van de kinderen met dit syndroom heeft problemen met ademhalen na de geboorte. Vaak ademen kinderen snel. Ook gebruiken kinderen hulpademhalingsspieren om voldoende ademhalen te halen. Dit is te zien als intrekken van de huid boven het borstbeen en tussen de ribben. Ook kunnen de neusvleugels meebewegen tijdens de ademhaling. Deze manier van ademhalen wordt respiratoire distress genoemd. Deze snelle manier van ademhalen is vermoeiend van een pasgeboren baby. Het kan daarom nodig zijn om de ademhaling tijdelijk te ondersteunen door middel van een beademingsapparaat.
Ook zijn kinderen gevoelig voor het krijgen van luchtweginfecties die kunnen zorgen voor een toename van problemen met ademhalen.

Tracheamalacie
Bij een deel van de kinderen beweegt de luchtpijp en de luchtwegen mee tijdens de ademhaling. Dit geeft een typisch "kakelend" geluid tijdens de ademhaling. Tijdens de uitademing wordt de luchtpijp heel nauw en kunnen beide kanten van de luchtpijp tegen elkaar aan vallen. De tracheamalacie zorgt ook voor ademhalingsproblemen. Met het ouder worden, wordt de luchtpijp groter, waardoor de luchtpijp minder snel samenvalt en de ademhalingsproblemen als gevolg van tracheamalacie minder worden.
Problemen met drinken
Een deel van de baby’s heeft problemen met drinken. Zij pakken de borst of de fles niet goed en laten deze gemakkelijk weer los. Vaak komt dit door de problemen met ademhalen, want een baby kan niet drinken en ademhalen tegelijk. De baby moet dus de borst of fles regelmatig los laten om weer adem te halen. Het voeden van baby’s met dit syndroom kost vaak meer tijd. Ook kunnen kinderen hierdoor licht van gewicht blijven. Dit wordt failure to thrive genoemd.

Uiterlijke kenmerken
Bij veel syndromen hebben kinderen vaak wat veranderde uiterlijke kenmerken. Hier hebben kinderen zelf geen last van, maar het kan de dokters helpen om te herkennen dat er sprake is van een syndroom en mogelijk ook van welk syndroom. Ook maakt dit vaak dat kinderen met hetzelfde syndroom vaak meer op elkaar lijken dan op hun eigen broertjes en zusjes, terwijl de kinderen toch niet familie van elkaar zijn.
Kinderen met het Chitayat-syndroom hebben vaak een afwijkende hoofdvorm, het hoofd is meer vierkant van vorm. Vaak hebben kinderen grote ogen die wat verder uit elkaar staan dan gebruikelijk. De neusbrug ligt vaak diep en de neuspunt staat wat omhoog gewipt. De meeste kinderen hebben volle lippen. De onderkaak is vaak wat kleiner dan de bovenkaak waardoor een overbeet ontstaat.
Craniosynostose
Een groot deel van de kinderen heeft een craniosynostose waardoor de schedel niet een ronde maar meer een vierkante vorm heeft. Hierdoor heeft ook het gezicht een andere vorm. Kinderen hebben hier zelf verder geen last van.
Handen en voeten
Vaak hebben kinderen met dit syndroom korte vingers en tenen. Vooral de middelvinger is vaak extra kort. De vingers of tenen kunnen naar de kant van de duim/grote teen of pink/kleine teen toegedraaid staan. Ook kunnen extra vingerkootjes gevonden worden aan de basis van de hand. Dit valt meestal alleen op wanneer een foto van de hand gemaakt wordt. Tussen de vingers kunnen kleine vliesjes gezien worden. De afwijkende stand van de voeten kan problemen geven met het vinden van passende schoenen.
Borstkas
Bij een deel van de kinderen heeft de borstkas een andere vorm dan gebruikelijk. De borstkas kan smaller van vorm zijn. Het borstbeen kan naar binnen toe staan, dit wordt een pectus excavatum genoemd.

Gevoeligheid voor infecties
Kinderen met dit syndroom zijn gevoelig voor het krijgen van infecties van de luchtwegen (bronchitis) of van de longen (longontsteking). Deze infecties verlopen ook heviger dan bij kinderen zonder dit syndroom. Sommige kinderen hebben tijdens een infectie opnieuw ondersteuning van een beademingsapparaat nodig.
Hoe wordt de diagnose Chitayat-syndroom gesteld?
Verhaal en onderzoek
Aan de hand van het verhaal van een kind met een ademhalingsproblemen op de babyleeftijd in combinatie met bijzondere uiterlijke kenmerken van het hoofd en de handen, kan worden vermoed dat er sprake is van een syndroom. Soms wordt direct herkend dat er sprake is van het Chitayat-syndroom, zeker wanneer deze aandoening in de familie voorkomt. Er zal aanvullend onderzoek nodig zijn om de definitieve diagnose te stellen.
Bloedonderzoek
Bij routine bloedonderzoek worden bij kinderen met dit syndroom geen bijzonderheden gevonden.
DNA-onderzoek
Tegenwoordig zal vaak door middel van een nieuwe genetische techniek (whole exome sequencing (WES)genoemd) deze diagnose gesteld kunnen worden zonder dat er specifiek aan gedacht was of naar gezocht is.
Foto van de handen en voeten
Een foto van de handen en voeten kan laten zien dat er sprake is van afwijkende aangelegde botten in de handen en in de voeten.
CT-scan
Door middel van een CT-scan kan bekeken worden of er sprake is van het te vroeg aan elkaar groeien van de schedelnaden.
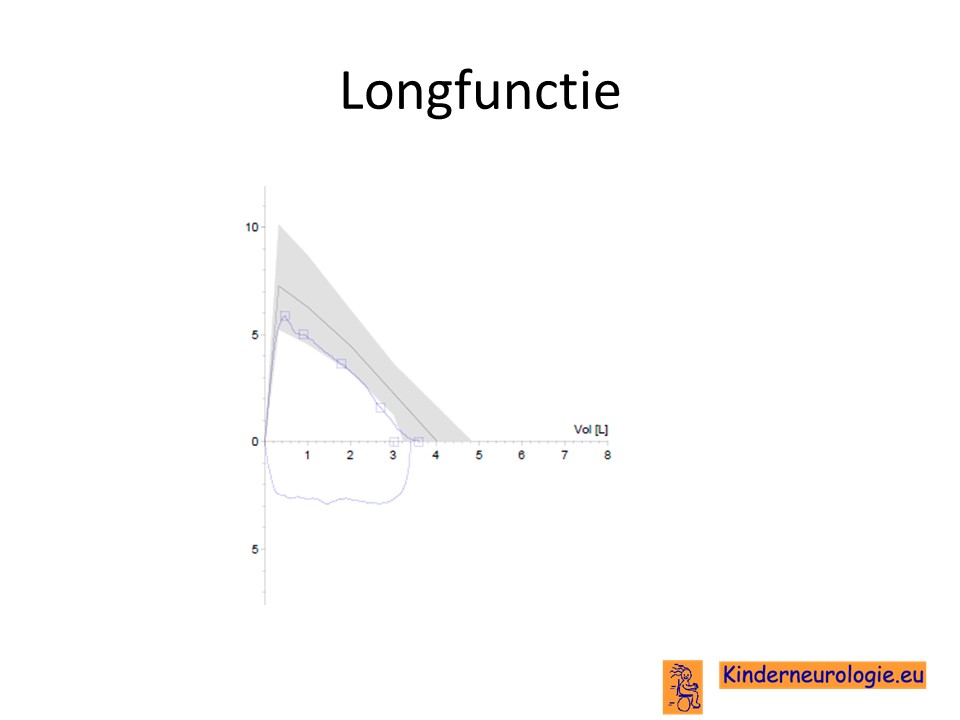
Longfunctie
Door middel van een longfunctieonderzoek kan bekeken worden hoe de longen functioneren. Vaak is de longfunctie bij kinderen met het Chityat syndroom verminderd.
Hoe wordt het Chitayat-syndroom behandeld?
Geen genezing
Er bestaat geen behandeling die het Chitayat-syndroom kan genezen. De behandeling is er op gericht om problemen zo vroeg mogelijk op te sporen en waar mogelijk te behandelen.
NICU
Een groot deel van de kinderen met het Chitayat syndroom heeft na de geboorte ademhalingsproblemen waarvoor kinderen opgenomen moeten worden in het ziekenhuis. Vaak worden kinderen opgenomen op een speciale afdeling voor pasgeborenen waar deze kinderen heel goed in de gaten gehouden kunnen worden. Een groot deel van de kinderen heeft enkele dagen tot weken ondersteuning door een beademingsapparaat nodig. Wanneer kinderen groter groeien en de luchtwegen groter worden, lukt het vaak om zonder het beademingsapparaat zelfstandig adem te halen. Er bestaat een risico op het ontstaan van een longonsteking, dan zullen antibiotica nodig zijn om de longontsteking te behandelen.
Sondevoeding
Een deel van de kinderen heeft tijdelijk sondevoeding nodig om te zorgen dat zij voldoende voedingsstoffen binnen krijgen. Moeders kunnen moedermelk afkolven, wat via de sonde gegeven kan worden. Het is ook mogelijk om kunstvoeding via de sonde te geven. De sonde is een slangetje die via de neus naar de maag toe loopt.

Kinderarts
In Nederland wordt door de zorg voor kinderen met een zeldzaam syndroom vaak gecoordineerd door de kinderarts in de eigen woonomgeving. Daarnaast kunnen kinderen ook begeleid worden door een speciale kinderarts die zich gespecialiseerd heeft in de zorg voor kinderen met een aangeboren en vaak zeldzame aandoeningen. Deze kinderarts heet kinderarts EAA: kinderarts voor erfelijke en aangeboren aandoeningen. In steeds meer ziekenhuizen in Nederland werken kinderartsen EAA. De kinderarts EAA stemt met de eigen kinderarts in de woonomgeving van het kind af hoe de zorg voor het kind zo optimaal mogelijk kan verlopen.

Fysiotherapie
Een fysiotherapeut kan ouders tips en adviezen geven hoe ze hun kind zo goed mogelijk kunnen stimuleren om er voor te zorgen dat de ontwikkeling zo optimaal als mogelijk verloopt. Ook kan een fysiotherapeut adviezen geven over tillen en verplaatsen.

Ergotherapie
De afwijkende stand van de handen kan het vastpakken van een potlood, pen of schaar lastiger maken. Er bestaan hulpmiddelen die het vasthouden van een pen en een potlood gemakkelijker kunnen maken. Vaak bestaan er ook speciale handenteams waar fysiotherapeuten samen met ergotherapeuten en revalidatieartsen advies geven.

Logopedie
Een logopediste kan tips en adviezen geven indien er problemen zijn met zuigen, drinken, kauwen of slikken. Sommige kinderen hebben baat bij een speciale speen (special need speen) waardoor het drinken uit de fles beter verloopt. Moeders kunnen borstvoeding kolven, zodat kinderen op deze manier toch borstvoeding als voeding kunnen krijgen via de fles.De logopediste kan ook adviezen geven hoe de mondspieren getraind kunnen worden, waardoor kinderen minder last hebben van kwijlen.

Diëtiste
Een diëtiste kan adviezen geven hoe kinderen voldoende voeding binnen kunnen krijgen om zich goed te voelen en voldoende te kunnen groeien.

Longarts
Kinderen met het Chitayat syndroom worden vaak vervolgd door de longarts om te kijken hoe hun longfunctie zich ontwikkelt en om te kijken of de longen ondersteund moeten worden met medicijnen om beter te kunnen functioneren. Sommige kinderen hebben baat bij pufjes die de luchtwegen wijder maken, bij anderen helpen deze helemaal niet.

Neurochirurg
Wanneer kinderen hinder hebben van de afwijkende vorm van de schedel, dan kan de neurochirurg door middel van een operatie de vorm van de schedel aanpassen.
Plastisch chirurg
Wanneer de stand van de vingers of tenen veel hinder geeft bij het uitvoeren van dagelijkse bezigheden kan een plastisch chirurg door middel van een operatie de stand van de vingers en tenen proberen te veranderen zodat kinderen deze beter kunnen gebruiken.
Antibiotica
Een deel van de kinderen die vaak terugkerende infecties heeft, heeft baat bij een lage dosering antibiotica om nieuwe infecties van de luchtwegen te voorkomen. Per kind moeten de voordelen van het geven van de antibiotica worden afgewogen tegen de nadelen ervan (antibiotica doden ook nuttige bacteriën in de darmen).

Financiële kant van zorg voor een kind met een beperking
De zorg voor een kind met een beperking brengt vaak extra kosten met zich mee. Er bestaan verschillende wetten die zorg voor kinderen met een beperking vergoeden.
Daarnaast bestaan regelingen waar ouders een beroep op kunnen doen, om een tegemoetkoming te krijgen voor deze extra kosten. Meer informatie hierover vindt u in de folder financiën kind met een beperking. .

Wat kun je als gezin zelf doen om om te gaan met het hebben van een aandoening bij een gezinslid?
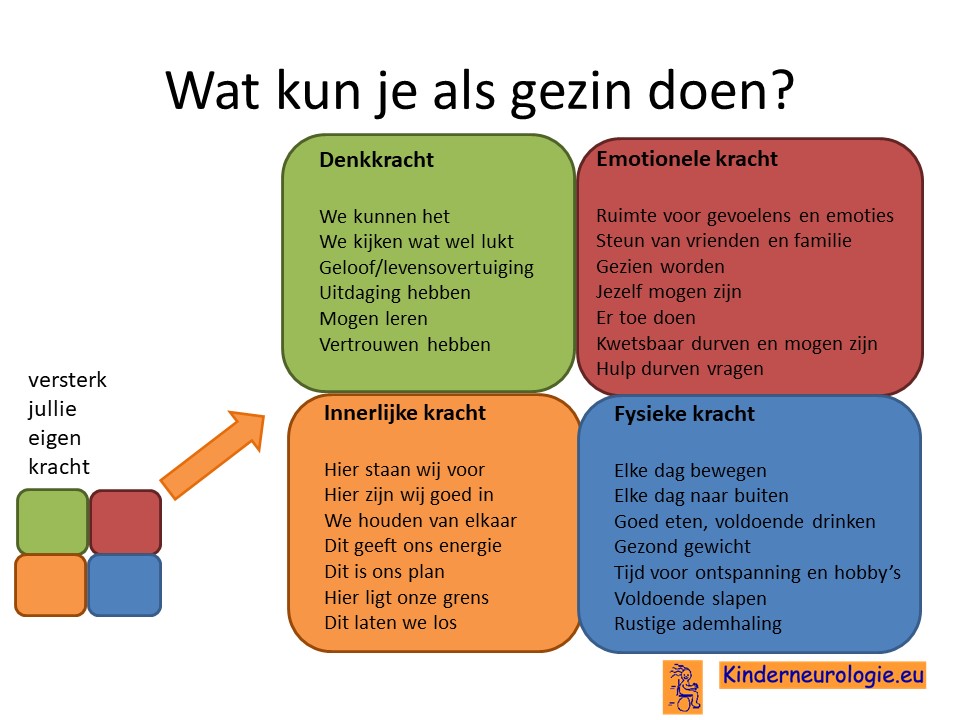
Als gezin van een kind waarbij er sprake is van een aandoening, is het goed om te zorgen dat jullie in de je kracht komen staan. Het is goed om te beseffen over welke denk-, emotionele-, innerlijke- en fysieke kracht jullie als gezin beschikken en hoe jullie deze kracht kunnen inzetten om goed voor ieder lid van het gezin te zorgen. Bekijk wat bij jullie als gezin past. Bekijk wat je kunt doen (of kunt laten) om deze kracht zo optimaal mogelijk in te zetten. En bedenk ook dat ieder lid van het gezin verschillende kwaliteiten heeft waarmee jullie elkaar kunnen aanvullen en kunnen versterken.
Begeleiding
Een maatschappelijk werkende of psycholoog kan begeleiding geven hoe het hebben van deze aandoening een plaats kan krijgen in het dagelijks leven. Het kost vaak tijd voor ouders om te verwerken dat de toekomstverwachtingen van hun kind er anders uit zien dan waarschijnlijk verwacht is.

Contact met andere ouders
Door het plaatsen van een oproepje op het forum van deze site site kunt u in contact komen met andere kinderen en ouders die bekend zijn met het Chitayat-syndroom.
Wat betekent het hebben van het Chitayat-syndroom voor de toekomst?
Verbeteren ademhalingsproblemen
Met het ouder worden, worden de luchtwegen groter en nemen de ademhalingsproblemen van kinderen met dit syndroom af. Wel blijven deze kinderen vaak gevoelige luchtwegen houden die zich gemakkelijk vernauwen (bronchospasme) en zijn kinderen vatbaar voor het krijgen van een ontsteking van de luchtpijp of de longen.
Transitie van zorg
Tussen de leeftijd van 16 en 18 jaar wordt de zorg vaak overgedragen van kinderspecialisten naar specialisten die de zorg aan volwassenen geven. Het is belangrijk om tijdig hierover na te denken. Is er behoefte de zorg over te dragen naar specialisten voor volwassenen of kan de huisarts de zorg leveren die nodig is.En als er behoefte is aan overdragen van de zorg naar specialisten voor volwassenen, naar welke dokter(s) wordt de zorg dan overgedragen? In welk ziekenhuis kan de zorg het beste geleverd worden. Het proces van overdragen van de zorg wordt transitie genoemd. Het is belanrgijk hier tijdig over na te denken en een plan voor te maken samen met de dokters die betrokken zijn bij de zorg op de kinderleeftijd. Ook verandert er veel in de zorg wanneer een jongere de leeftijd van 18 jaar bereikt. Voor meer informatie over deze veranderingen verwijzing wij u naar het artikel veranderingen in de zorg 18+
![]()
Levensverwachting
Er is weinig bekend over volwassenen met dit syndroom. Naar verwachting zal de levensverwachting niet anders zijn dan van andere kinderen zonder dit syndroom wanneer kinderen de eerste fase na de geboorte met ademhalingsproblemen goed doorgekomen zijn.
Kinderen krijgen
Er zijn geen aanwijzingen dat het hebben van dit syndroom van invloed is op de vruchtbaarheid. Kinderen van een ouder met het Chitayat-syndroom hebben 50% kans om zelf ook dit syndroom te krijgen.
Kinderen hoeven niet dezelfde klachten en dezelfde mate van ernst van de klachten te krijgen als hun ouder. Kinderen kunnen minder klachten hebben dan hun ouder, maar ook juist meer klachten. Dit valt van te voren niet goed te voorspellen. Indien de volwassene geen kinderen wil of kan krijgen, moet wellicht nagedacht moeten worden over anticonceptie, waarover u in deze folder meer informatie vindt.
Hebben broertjes en zusjes een vergrote kans om ook het Chitayat-syndroom te krijgen?
Het Chitayat-syndroom is een erfelijke aandoening. Vaak is de fout in het erfelijk materiaal bij het kind zelf is ontstaan. De kans is dan heel klein dat broertjes en zusjes ook deze aandoening krijgen. Dit zou alleen het geval kunnen zijn als de fout bij de moeder in de eicellen of bij de vader in de zaadcellen zou zitten, zonder dat de fout ergens anders in het lichaam van deze ouder zit. Dit wordt ouderlijk mocaisisme genoemd. De kans hierop is klein.
Wanneer een van de ouders zelf het
Chitayat-syndroom heeft, dan is de kans dat een kind dit syndroom krijgt 50%.
Een klinisch geneticus kan hier meer informatie over geven.

Prenatale diagnostiek
Door middel van een vlokkentest in de 12e zwangerschapsweek of een vruchtwaterpunctie in de 16e zwangerschapsweek bestaat de mogelijkheid om tijdens een zwangerschap na te gaan of een broertje of zusje ook deze aandoening heeft. Beide ingrepen hebben een klein risico op het ontstaan van een miskraam (0,5% bij de vlokkentest en 0,3% bij de vruchtwaterpunctie). De uitslag van deze onderzoeken duurt twee weken. Voor prenatale diagnostiek kan een zwangere de 8ste week verwezen worden door de huisarts of verloskundige naar een afdeling klinische genetica. Meer informatie over prenatale diagnostiek kunt u vinden op de website: www.pns.nl
Wilt u dit document printen dan kunt u hier een pdf-versie downloaden.
Wilt u ook uw verhaal kwijt, dat kan: verhalen kunnen gemaild worden via info@kinderneurologie.eu en zullen daarna zo spoedig mogelijk op de site worden geplaatst. Voor meer informatie zie hier.
Heeft u foto's die bepaalde kenmerken van deze aandoening duidelijk maken en die hier op de website mogen worden geplaatst, dan vernemen wij dit graag.
Links
www.zeldsamen.nl
(netwerken voor zeldzame genetische syndromen)
Referenties
1.Chitayat syndrome: hyperphalangism, characteristic facies, hallux valgus and bronchomalacia results from a recurrent c.266A>G p.(Tyr89Cys) variant in the ERF gene. Balasubramanian M, Lord H, Levesque S, Guturu H, Thuriot F, Sillon G, Wenger AM, Sureka DL, Lester T, Johnson DS, Bowen J, Calhoun AR, Viskochil DH; DDD Study, Bejerano G, Bernstein JA, Chitayat D. J Med Genet. 2017;54:157-165.
2. Variable pulmonary manifestations in Chitayat syndrome: Six additional affected individuals. Suter AA, Santos-Simarro F, Toerring PM, Abad Perez A, Ramos-Mejia R, Heath KE, Huckstadt V, Parrón-Pajares M, Mensah MA, Hülsemann W, Holtgrewe M, Mundlos S, Kornak U, Bartsch O, Ehmke N. Am J Med Genet A. 2020;182:2068-2076
Laatst bijgewerkt: 10 juli 2022 voorheen: 2 juni 2021
Auteur: JH Schieving
Heeft uw kind nog andere symptomen, laat het ons weten.